Flat bottomed potentials (FBPs) are a unique type of restraint we can apply to specific particles in a molecular dynamics simulation.
In short, we define a fixed geometric region within the simulation box to which certain atoms will either be attracted or repelled.
They're also a little tricky to understand from a technical perspective.
In this post, I'll briefly discuss the maths, how to implement an FBP in the topology and restraints files, and finish up with some practical notes for usage.
The Maths
As we can see from the GROMACS manual, the general equation for an FBP is given as: $$ V_{fb}(r_i) = \frac{1}{2}k_{fb}[d_g(r_{i};R_{i}) - r_{fb}]^2 H[d_g(r_{i};R_{i}) - r_{fb}] $$ with the most important parameters being:
- $d_g(r_{i};R_{I})$, the geometry of the shape we wish to define (FBPs can be spheres, cylinders, or layers, see the GROMACS manual for more info on the shapes).
- $r$, the radius of our shape. A negative radius keeps atoms outside of our FBP region, a positive radius keeps them within.
- $k$, the force constant applied to the chosen atoms when they are not inside or outside of our chosen shape.
- $H$, the Heaviside step function which "turns on" the force constant once a restrained atom leaves the FBP region.
Now, let's have a look at an image from the GROMACS manual:
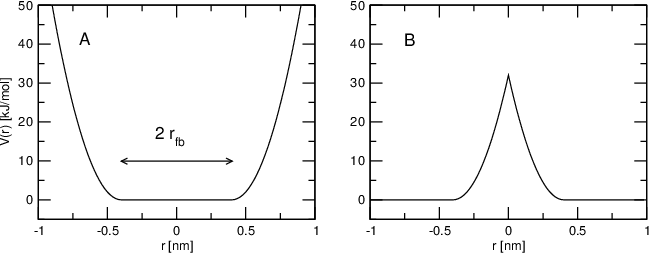
Figure A shows a "positive" (non-inverted) FBP, which keeps a chosen atom within a certain shape, by applying a force of $k$ if the restrained particle strays outside of the radius $r_{fb}$.
Figure B shows a "negative" (inverted) FBP, which instead keeps the particle away from the shape.
Implementation
Now, what makes FBPs in GROMACS rather annoying from a technical perspective is the need to split the definition across two files:
1. a section in the .itp topology file defining the shape, radius, force constant, as well as the specific atoms to be restrained.
2. a restraints.gro file which contains the x, y, and z coordinates of our FBP region for a given atom
To show how the two work together, consider this example from one of my previous projects (GitHub, Publication), in which we use FBPs to define a pore region in a coarse-grained (Martini 3) membrane tubule, to stop lipids from passing through the pore.

Topology File
To make these pores, I wanted two cylindrical FBPs crossing the box, one in x, one in y. By defining a negative radius of -2.5nm, I'm keeping the restrained molecules out of the FBP geometries. And I wanted a strong force constant, k=5000 (where k=$KJ \cdot mol^{-1}\cdot nm^{-2}$).
I define all of these in the itp file for the molecule I want to restrain (here, my coarse-grained phospholipid).
#ifdef POSRES_PL
; Flat-bottomed position restraint for each PL
[ position_restraints ]
; numatoms functype g r k
; (nm) (kJ mol−1nm−2)
05 2 6 -2.5 5000
06 2 6 -2.5 5000
07 2 6 -2.5 5000
08 2 6 -2.5 5000
09 2 6 -2.5 5000
10 2 6 -2.5 5000
11 2 6 -2.5 5000
12 2 6 -2.5 5000
05 2 7 -2.5 5000
06 2 7 -2.5 5000
07 2 7 -2.5 5000
08 2 7 -2.5 5000
09 2 7 -2.5 5000
10 2 7 -2.5 5000
11 2 7 -2.5 5000
12 2 7 -2.5 5000
#endif
Where the columns correspond to:
-
The atom number within the molecule I'm restraining.
-
The function type: here, we put a 2 for all entries, which is the function type for FBPs under the
[position_restraints]directive. -
The shape (g) of the FBP. Here, I'm using
6for a cylinder spanning the x-dimension, and7for a cylinder spanning the y-dimension. -
The radius $r$ of the FBP.
-
The force constant $k$.
This is a nice example, as we can see that we can define multiple FBPs on the same particle.
Restraints File
However, you may notice that we haven't yet centered the FBP anywhere! This is where the restraints.gro file comes in.
A snippet from my restraints.gro file looks like this:
Expect a large membrane in water
71260
1POPC NC3 1 14.799 14.799 05.000
1POPC PO4 2 14.799 14.799 05.000
1POPC GL1 3 14.799 14.799 05.000
1POPC GL2 4 14.799 14.799 05.000
1POPC C1A 5 14.799 14.799 05.000
...
55975W W75687 10.673 25.982 5.496 0.0510 0.0486 -0.2671
55976W W75688 22.924 23.116 3.672 0.0720 0.0694 -0.1156
29.59805 29.59805 10.00000
Since I'm restraining the POPC lipids, I define the FBP center of geometry on the POPC particles/atoms as x,y,z in the gro file coordinates.
Since I'm not restraining the water (W) molecules, they can simply be left as is.
On my github I have a script that will take care of this, which would generate a restraints file from your starting structure file, which would be run as:
python gen_gromacs_restraints.py -c $INPUT_GRO -r POPC -r POPE -x 14.799 -y 14.799 -z 5
Some practical notes
- A given particle can have multiple FBPs placed upon it, but they all must come from the same set of coordinates as found in the
restraints.gro. - A poorly placed flat-bottomed potential will probably cause your system to explode immediately. If a system suddenly has a force of 5000 kJ/mol/nm^2 applied to every molecule in a given region, don't expect it to respond kindly.
- As such, be generous with the radius and soft with the force constant, at least at first. It may be a good practice to "grow" your FBP by gradually increasing $r$ and $k$ over a few subsequent simulations. Be kind to your simulations!
- You can check the
.logfile to make sure your FBP is actually running; there should be an entryFlat-b. P-R.. If it shows a 0.00000, this means that no particles are currently being affected by the FBPs. - If an FBP has been defined in the topology, it will not run without a restraints.gro file, which is flagged at the
gmx gromppstep with-r. If you're not sure if you've got it working properly, trygrompp-ing without-r.